af Viggo Andreasen, IMFUFA
Mange alvorlige infektionssygdomme påfører den berørte befolkning en høj dødelighed og har derfor gennem naturlig selektion stor indflydelse på værts-befolkningens genetiske sammensætning. Imidlertid er den teoretiske forståelse af fænomenet ringe, specielt sammenhængen mellem sygdommens hyppighed og værternes genetiske struktur er vanskelig at beskrive. Sammen med Freddy Christiansen, en kollega fra Århus Universitet, har jeg arbejdet på at komme lidt længere i beskrivelsen af dette klassiske problem i matematisk biologi.
Den mest kendte vekselvirkning mellem sygdom og genetik findes i malaria-inficerede områder af Afrika. Her bærer op til 30\% af befolkningen et gen, S-genet, som forårsager en defekt i de røde blodlegemer. Individer med to kopier af S-genet dør i en tidlig alder, mens heterozygoter, altså individer med et normalt gen og et S-gen, lider af en let men kronisk iltmangel, "segl-celle anæmi". Heterozygoter er imidlertid delvist beskyttet mod den farlige malaria plasmodium falsiparum. Hyppigheden af S-genet holdes i skak af to modsatrettede faktorer i en såkaldt beskyttet polymorfi: hvis S-genet er hyppigt, vil mange af heterozygoternes børn fødes med to S-gener og dø tidligt. Dette vil reducere hyppigheden af S-genet. Hvis genet er sjældent, vil bærere af S-genet være beskyttet mod malaria, og deres børn får højst et S-gen. S-genet vil derfor vokse i hyppighed. Malaria-segl-celle systemet blev beskrevet i 60erne, og i dag kender man S-genets molekylære struktur såvel som den fysiologiske mekanisme, der beskytter eterozygoter mod malaria. Derimod ved vi ikke, hvordan størrelsen af malarias sygdomstryk påvirker hyppigheden af S-genet. I Sydøstasien findes et lignende malaria-beskyttende gen, og det er fornylig blevet foreslået, at cystisk fibrose oprindelig er opstået som en tilsvarende beskyttelse mod tyfus. I såvel plante- som dyreverdenen findes flere eksempler på infektionssygdomme, der påvirker værtens genetiske struktur.
Inspirationen til mit samarbejde med Freddy Christiansen kom fra to kilder. Nogle år før vi begyndte vores arbejde, havde den amerikanske matematiker Karen Beck udviklet en model af sygdomsspredningen, hvor hver genotype blev fulgt særskildt. Modellen var biologisk set nem at forstå, men den matematiske struktur var meget kompliceret, idet den bestod af 9 koblede differentialligninger. Beck's formulering forudsatte, at det betragtede gen havde en lille virkning på værtens overlevelse. Hun kunne nu udnytte denne lille effekt til at betragte modellen som et såkaldt {\it singulært pertubationsproblem} og dermed bestemme den langsomme ændring i genhyppighed. Hendes analyse var vanskelig at gennemskue, bl.a. fordi de omfattende beregninger kun kunne udføres ved computerprogrammer til symbolsk algebra.
Den anden inspirationskilde var et tidligere arbejde af Freddy Christiansen, hvor han havde bemærket, at Wrights fikseringsindex F havde pæne dynamiske egenskaber. Indexet F benyttes normalt som et statistisk mål for, hvor meget populationen afviger fra den velkendte Hardy-Weinberg ligevægt, så det var bemærkelsesværdigt, at F skulle være velegnet som dynamisk variabel.
Vores ide var nu at transformere Beck's model til et koordinatsystem bestående af total populationsstørrelse, genhyppighed samt indexet F og derefter undersøge det singulære perturbationsproblem i dette koordinatsystem. Lidt overraskende fandt vi, at denne transformation havde en række pæne matematiske egenskaber, som gjorde beregningerne simple og gennemskuelige. Til slut kunne vi ved en simpel udgave af Becks metoder bestemme den langsomme dynamik i genhyppigheden. Dermed kunne vi i detaljer beskrive, hvordan genhyppigheden påvirkes af sygdomshyppigheden samt den genetiske varition i bl.a. overlevelse og modtagelighed.
Som opfølgning har vi studeret en model, hvor såvel værtspopulationen som sygdommen har genetisk betinget variation. Den resulterende langsomme dynamik kan her være ganske kompliceret. For passende valg af parametre kan systemet bringes til at svinge - svarende til de såkaldte gen-for-gen systemer, der er kendt fra sygdoms-resistens hos flere afgrøder. Imidlertid viser en nøjere matematisk undersøgelse (bifurkationsanalyse), at modellen i denne situation er strukturel ustabil hvilket vil sige, at gensammensætningen vil afhænge af detaljer, som i første omgang ikke er inkluderet i modellen.
Vores arbejde har ikke givet et fuldstændigt svar på, hvordan infektionssygdomme påvirker den naturlige selektion. Modellen og metoden skal opfattes som et bidrag til udviklingen af det sprog, man har til rådighed, når man arbejder med denne type processer.
Referencer
Andreasen, V. and Christiansen, F.B. (1993). Disease-induced natural selection
in a diploid host. Theor. Pop. Biol, 44: 261--298.
Andreasen, V. and Christiansen, F.B.\ (1995). Slow co-evolution of a viral pathogen and its diploid host. Phil. Trans. Roy. Soc. London, B348: 341--354.
V.Andreasen.
Natural selection in diploid hosts exposed to an infectious disease.
In U.Dieckmann, J.A.J. Metz, M.W. Sabelis, and K.Sigmund, editors, The
Adaptive Dynamics of Infectious Diseases: In Pursuit of Virulence Management.
Cambridge University Press, Cambridge, New York, 2001.
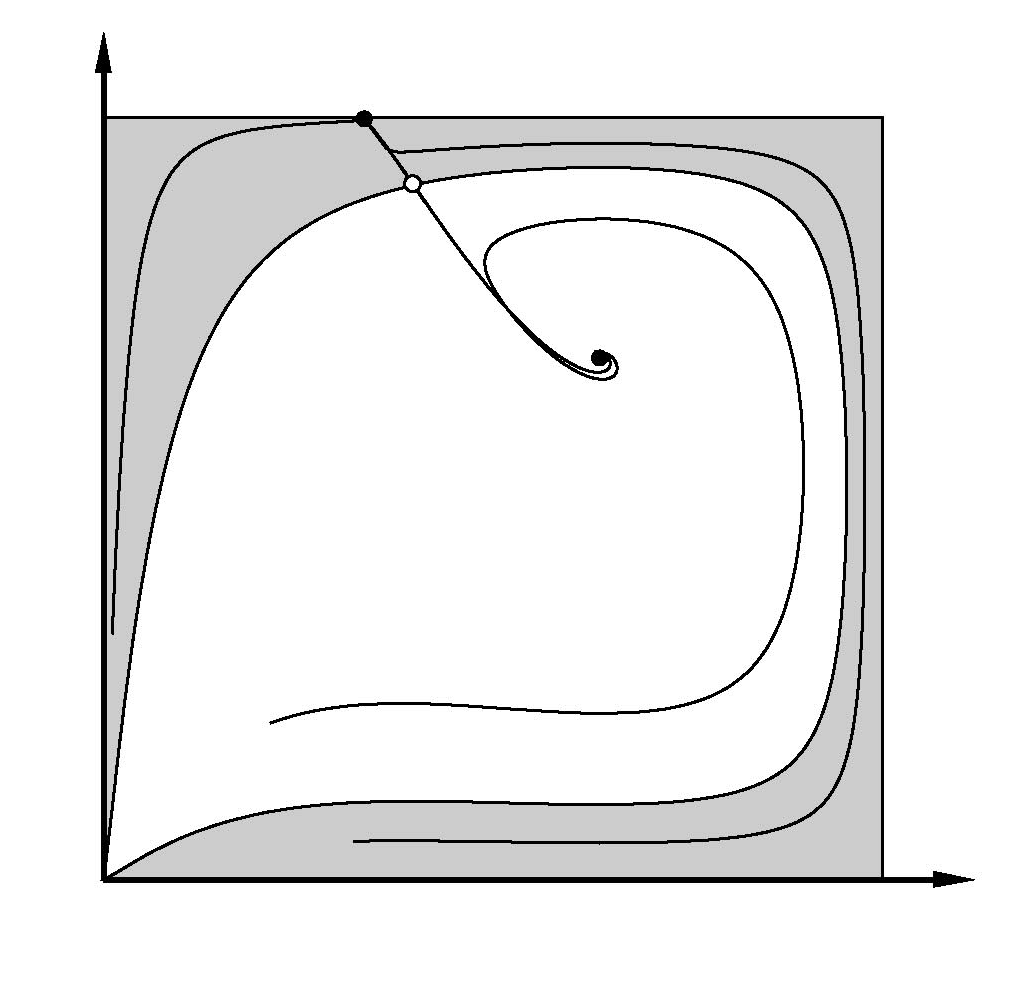
I artiklen omtales en model, der beskriver sammenhængen mellem en dødelig infektionssygdom, der forekommer i to varianter, og værtsbefolkningens genetiske sammensætning. Figuren viser hvordan hyppigheden p af et bestemt gen i værtspopulationen og hyppigheden af en bestemt sygdomsvariant (pi) ∏ kan udvikle sig gennem tiden (angivet med pile). For de benyttede parameterværdier kan gensammensætningen bevæge sig mod to forskellige ligevægte o afhængigt af sammensætningen af gener på starttidspunktet.